微生物測(cè)序
微生物組(Microbiome)是指一個(gè)特定環(huán)境或生態(tài)系統(tǒng)中全部微生物及其遺傳信息的集合,?蘊(yùn)藏著極為豐富的微生物資源。全面系統(tǒng)地解析微生物組的結(jié)構(gòu)和功能,?將為解決人類(lèi)面臨的能源、生態(tài)環(huán)境、工農(nóng)業(yè)生產(chǎn)和人體健康等重大問(wèn)題帶來(lái)新思路
細(xì)菌完成圖(三代測(cè)序)
基因組是闡釋生命現(xiàn)象和揭示生命規(guī)律的重要手段,三代PacBio HiFi測(cè)序模式可產(chǎn)生既兼顧長(zhǎng)讀長(zhǎng),又具有高精度的測(cè)序結(jié)果,貝瑞基因HiFi細(xì)菌完成圖測(cè)序,即利用PacBio HiFi測(cè)序模式對(duì)細(xì)菌物種進(jìn)行基因組de novo組裝,從而獲得該細(xì)菌種完整基因組序列,該技術(shù)大大提升了細(xì)菌基因組組裝的完整性和準(zhǔn)確性,可真正實(shí)現(xiàn)細(xì)菌0 gap組裝。
真菌精細(xì)圖(三代測(cè)序)
三代真菌精細(xì)圖,即采用PacBio測(cè)序平臺(tái),利用高深度三代數(shù)據(jù)進(jìn)行真菌基因組de novo組裝,獲得真菌基因組序列信息的一種技術(shù)手段。真菌基因組一般在10M-150M,有多核和雜合等現(xiàn)象,利用三代測(cè)序技術(shù)的優(yōu)勢(shì)破譯真菌基因組,能克服二代測(cè)序?qū)Ω逩C、高重復(fù)、高雜合位置檢出不準(zhǔn)確的現(xiàn)象,得到更加完整和均勻的基因組,結(jié)合HiC輔助組裝技術(shù)可獲得染色體水平全基因組序列圖譜,此外,測(cè)序同時(shí)還可獲得甲基化修飾位點(diǎn)信息。
全長(zhǎng)16s(三代測(cè)序)
常規(guī)二代16S僅針對(duì)單個(gè)可變區(qū)或者連續(xù)兩到三個(gè)可變區(qū)進(jìn)行測(cè)序,而基于PacBio HiFi測(cè)序的全長(zhǎng)16S(V1-V9)可獲取微生物16S的全長(zhǎng)序列,突破了二代測(cè)序讀長(zhǎng)較短的局限性,能夠更好的實(shí)現(xiàn)微生物在種水平上的分類(lèi), 可獲得物種分類(lèi)、豐度、種群結(jié)構(gòu)等更加深入和全面的信息。基于PacBio HiFi測(cè)序技術(shù)的全長(zhǎng)16s測(cè)序?yàn)槲⑸锒鄻有匝芯刻峁┝藦?qiáng)有力的技術(shù)手段。
宏基因組(二代測(cè)序)
二代宏基因組測(cè)序技術(shù)是依托二代測(cè)序平臺(tái)以特定環(huán)境中的整個(gè)微生物群落作為研究的對(duì)象,不需要對(duì)微生物進(jìn)行分離培養(yǎng),提取環(huán)境微生物總DNA進(jìn)行研究,進(jìn)行微生物群體的物種分類(lèi)、復(fù)雜度分析、群落結(jié)構(gòu)、功能注釋、樣品間的物種或基因差異及物種間的代謝網(wǎng)絡(luò)研究。與傳統(tǒng)微生物研究方法相比,宏基因組測(cè)序技術(shù)規(guī)避了絕大部分微生物不能培養(yǎng)、痕量菌無(wú)法檢測(cè)的缺點(diǎn)。
應(yīng)用方向
- 精準(zhǔn)醫(yī)療:探究腸道、皮膚、陰道等菌群環(huán)境對(duì)疾病的影響,尋找與代謝類(lèi)疾病、消化類(lèi)疾病、自身免疫性疾病等相關(guān)的biomarker及與癌癥相關(guān)的治療靶點(diǎn);
- 藥物代謝:探究功能食品、中藥的體內(nèi)代謝及藥效機(jī)制;
- 傳統(tǒng)發(fā)酵生產(chǎn):關(guān)鍵風(fēng)味物質(zhì)溯源及控制發(fā)酵微生物菌種優(yōu)化改良,及特殊功能菌株、基因挖掘、工程菌開(kāi)發(fā)等;
- 畜牧漁養(yǎng)殖:研究腸道、瘤胃等微生物與動(dòng)物繁殖、生長(zhǎng)發(fā)育、營(yíng)養(yǎng)健康、免疫和疾病治療等的互作關(guān)系、對(duì)環(huán)境的響應(yīng)及對(duì)生產(chǎn)的影響;
- 大農(nóng)業(yè):研究根系微生物對(duì)植物生長(zhǎng)、抗逆的影響,指導(dǎo)種植方式;
- 環(huán)境領(lǐng)域:研究特定環(huán)境下微生物研究,解決有機(jī)肥發(fā)酵處理、污水治理、石油降解、水體及海洋環(huán)境等環(huán)境相關(guān)問(wèn)題;
案例解析
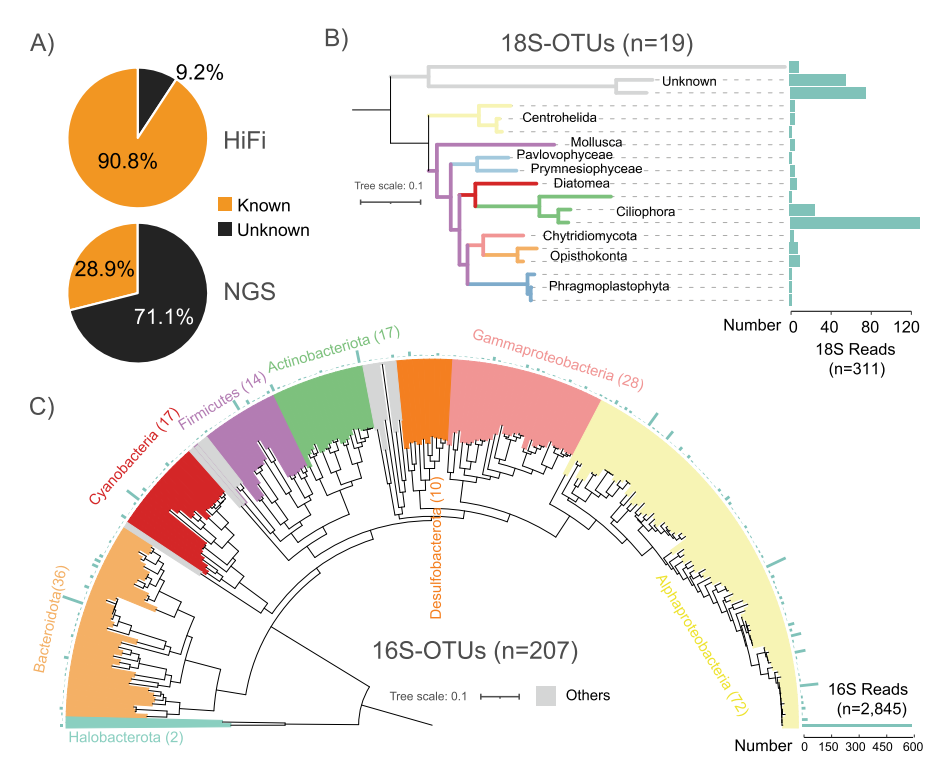
基于HiFi reads的分類(lèi)分辨率。(A) HiFi和NGS讀數(shù)具有(已知)/不具有(未知)分類(lèi)信息的百分比。(B和C)基于19個(gè)全長(zhǎng)18S-OTUs (B)和207個(gè)全長(zhǎng)16S-OTUs (C)的系統(tǒng)進(jìn)化樹(shù)
利用HiFi宏基因組測(cè)序技術(shù)改進(jìn)西藏鹽湖沉積物中宏基因組組裝和病毒組裝
該文通過(guò)應(yīng)用長(zhǎng)讀長(zhǎng)測(cè)序技術(shù)和短讀長(zhǎng)測(cè)序技術(shù)對(duì)青藏高原鹽湖沉積物樣品進(jìn)行測(cè)序,評(píng)估了不同組裝策略在宏基因組組裝和病毒組裝中的性能。28.9% 的 NGS 讀取可以分配到已知的分類(lèi)等級(jí),而HiFi數(shù)據(jù)的讀取分配率為90.8%,大約是NGS的三倍。HiFi reads的物種級(jí)別分配率為67.19%,而短讀長(zhǎng)的相應(yīng)比率僅為5.59%,說(shuō)明HiFi reads在分類(lèi)方面的強(qiáng)大能力。總共鑒定出17個(gè)最小相對(duì)豐度大于0.1%的門(mén),大大超過(guò)了NGS技術(shù)的檢索性能。這為復(fù)雜環(huán)境樣品的物種組成研究,特別是對(duì)真核微生物的了解提供了便利。
參考文獻(xiàn):Tao Y, Xun F, Zhao C, Mao Z, et al. Improved Assembly of Metagenome-Assembled Genomes and Viruses in Tibetan Saline Lake Sediment by HiFi Metagenomic Sequencing. Microbiol Spectr. 2023 Feb 14;11(1):e0332822.
北京市昌平區(qū)科技園區(qū)生命園路4號(hào)院5號(hào)樓
客服熱線:400-610-8005

